AFI Terra Pipeline
1. Overview
The afi_terra pipeline detects and identifies Rickettsiales organisms — primarily Orientia, Rickettsia, and related genera — from 16S rRNA metagenomic sequencing data. It runs on Terra, the Broad Institute's cloud execution platform, using WDL 1.0 workflows backed by Docker images.
Target organisms
The pipeline is designed to detect genera in the order Rickettsiales. For Orientia and Rickettsia specifically, a confirmatory 16S rRNA alignment step provides higher specificity. All other genera detected by the classifier (e.g., Leptospira, Burkholderia, Anaplasma) are reported from Centrifuger read counts alone.
Two workflows
| Workflow | WDL file | Use case |
|---|---|---|
AFI_16S_Main | wdl/AFI_16S_Main.wdl | Single sample |
AFI_16S_Batch | wdl/AFI_16S_Batch.wdl | Full sequencing run (scatter/gather) |
For clinical use, run the batch workflow. It processes all samples in a run together, automatically computing a per-run NTC background, and producing a consolidated run_summary.tsv. The Terra Sheet Builder desktop app (Section 8) generates the required sample sheet TSV without manual editing.
Two sample modes
| Mode | Use case | Primary output |
|---|---|---|
routine | Clinical samples with unknown organisms | routine_summary.tsv |
validation | Samples with known expected organisms | validation_summary.tsv |
A single batch run can contain both modes simultaneously.
2. Architecture and Data Flow
Processing steps
Each sample passes through up to nine steps. Steps 1–5 run in Phase 1; steps 6–8 run in Phase 2 after per-run NTC backgrounds are computed; step 9 is the final batch-level gather.
Step 1 Human read dehosting NCBI SRA Scrubber (optional, default on)
Step 2 QC / adapter trimming fastp v0.23.4
Step 3 Taxonomic classification Centrifuger -> kreport -> genus counts TSV
Step 4 16S alignment minimap2 -> BAM
Step 5 Alignment metrics extract_rick16s_metrics.py -> align_metrics.tsv
---- batch gather: BuildNTCBackground (NTC/NC samples only; per run_id) ----
---- batch gather: MatchNTCBackground (assigns each sample its run's NTC) ----
Step 6 NTC-aware interpretation call_taxa.py -> calls.tsv + taxa_evidence.tsv
Step 7a Validation summary compare_expected (validation mode only)
Step 7b Routine summary summarize_routine (routine mode only)
Step 8 Run summary run_summary.tsv (batch only)
Batch workflow phases
Phase 1 scatter — runs steps 1–5 for all samples in parallel.
BuildNTCBackground — collects all NTC/NC metrics and computes one ntc_background_<run_id>.tsv per distinct run_id.
MatchNTCBackground — maps each sample to the NTC background for its run_id. This task fails if any run_id has no NTC/NC sample.
Phase 2 scatter — runs steps 6–7 for all samples using matched NTC backgrounds.
BuildRunSummary — merges all per-sample summaries into a single run_summary.tsv.
3. Reference Files and Databases
16S Rickettsiales panel
| File | Description |
|---|---|
rickettsiales_panel_16S.fa | 16S reference sequences, original |
rickettsiales_panel_16S.clean.fa | Cleaned version (recommended) |
rickettsiales_panel_16S.mmi | Pre-built minimap2 index |
rickettsiales_panel_16S.clean.fa or the pre-built .mmi index. The pre-built index avoids index-build overhead at runtime.Centrifuger database
The database (~67 GiB compressed) is stored in GCS as a TAR.GZ archive. Provide two inputs:
| Input | Example | Description |
|---|---|---|
centrifuger_db | centrifuger_bact_arch_plus_rickettsiales | Index prefix string |
centrifuger_db_archives | ["gs://bucket/...tar.gz"] | Array of GCS paths |
centrifuger_memory = "96G" and centrifuger_disks = "local-disk 375 HDD". The default Terra VM (1 CPU / 2 GB RAM) is far too small and will cause the task to fail.4. Importing Workflows into Terra
Option 1: Dockstore (recommended)
- In Dockstore, link the GitHub repository
PHemarajata/afi_terra. - Create or refresh a workflow version from the desired Git tag or branch.
- In Terra, navigate to Workflows and click Find a Workflow.
- Select Dockstore as the source and import via TRS. Terra resolves all imports automatically.
Option 2: Direct ZIP upload
If Dockstore is not available, create a ZIP preserving relative directory paths:
zip -r afi_terra_wdl.zip \ wdl/AFI_16S_Batch.wdl \ wdl/AFI_16S_Main.wdl \ wdl/tasks/ \ NCBI_scrub_PE/tasks/quality_control/read_filtering/task_ncbi_scrub.wdl
AFI_16S_Batch.wdl alone will fail. It imports AFI_16S_Main.wdl and all task WDLs via relative paths.5. Running the Single-Sample Workflow
ntc_background.tsv to be supplied manually.Required inputs
| Input | Type | Description |
|---|---|---|
sample_id | String | Unique sample identifier |
r1_fastq | File | GCS path to R1 FASTQ (.fastq.gz) |
r2_fastq | File | GCS path to R2 FASTQ (.fastq.gz) |
rickettsiales_panel | File | GCS path to 16S reference FASTA or .mmi index |
ntc_background | File | GCS path to NTC background TSV; use placeholder for first pass |
centrifuger_db | String | Centrifuger index prefix |
centrifuger_db_archives | Array[File] | GCS paths to TAR.GZ archives |
Mode and type inputs
| Input | Default | Allowed values |
|---|---|---|
mode | routine | routine, validation |
sample_type | clinical | NTC, NC, PC_MIX8, PC_SINGLE, MIXED4, clinical, PC |
use_human_scrub | true | true, false |
Launching in Terra
- Navigate to Workflows and select
AFI_16S_Main. - Choose Run workflow with inputs defined by file paths.
- Upload or paste your input JSON, then click Run Analysis.
6. Running the Batch Workflow
The batch workflow (AFI_16S_Batch) processes one or more sequencing runs in a single Terra submission. It scatters each sample through processing, automatically builds a per-run NTC background, and produces a consolidated run summary.
run_id matching its run; NTC backgrounds are computed independently per run_id.Per-sample arrays
| Input | Type | Description |
|---|---|---|
run_ids | Array[String] | Sequencing run identifier per sample |
sample_ids | Array[String] | Sample identifiers |
r1_fastqs | Array[File] | R1 FASTQ GCS paths |
r2_fastqs | Array[File] | R2 FASTQ GCS paths |
sample_types | Array[String] | Sample type for each sample |
modes | Array[String] | routine or validation per sample |
expected_taxa | Array[String] | Expected taxa string; "" for routine samples |
Sample types in a batch run
| sample_type | Description | Contributes to NTC background |
|---|---|---|
NTC | No-template control | Yes |
NC | Negative control (alias for NTC) | Yes |
PC_MIX8 | 8-organism positive control | No |
PC_SINGLE | Single-organism positive control | No |
clinical | Clinical patient sample | No |
PC | Generic positive control | No |
run_id in the batch must have at least one sample with sample_type of NTC or NC; otherwise MatchNTCBackground will fail.7. The Two-Pass Pattern
The batch workflow handles NTC backgrounds entirely automatically — no manual intervention is needed. The two-pass procedure below applies only to the single-sample workflow or exceptional reprocessing scenarios.
Step 1: First pass — placeholder NTC background
Supply wdl/inputs/ntc_background.placeholder.tsv (all-zero thresholds) as ntc_background. Collect the align_metrics output for each NTC/NC sample from Terra.
Step 2: Build run-specific NTC background
python3 scripts/build_ntc_background_from_metrics.py \ --ntc-metrics path/to/NTC_S11.align_metrics.tsv \ --ntc-metrics path/to/NTC_S12.align_metrics.tsv \ --out wdl/inputs/run3.ntc_background.tsv
Step 3: Upload and re-run
gsutil cp wdl/inputs/run3.ntc_background.tsv gs://YOUR_BUCKET/ref/run3_ntc_background.tsv
Re-submit the sample with ntc_background pointing to the real file. The second pass produces NTC-corrected calls.
8. Terra Sheet Builder (GUI)
AFI_16S_Batch without manually editing JSON or TSV files. It is the recommended way to prepare batch inputs for routine clinical use.
Getting the application
Pre-built binaries are built automatically by GitHub Actions. Download the artifact for your platform from Actions → Build Terra Sheet Builder on the repository page:
| Platform | Artifact |
|---|---|
| Linux (amd64) | Single-file ELF executable |
| Windows (x64) | .exe, no console window |
| macOS Intel | .app bundle, zipped |
| macOS Apple Silicon (arm64) | .app bundle, zipped |
Run from source (requires Python 3.10+):
pip install PySide6 python3 tools/terra_sheet_builder/terra_sheet_builder.py
Step-by-Step Walkthrough
The following walkthrough covers generating a Terra metadata sheet for the AFI_16S_Batch workflow for a two-run batch.
Specify the number of runs in this batch
On the first screen, enter how many sequencing runs you want to process together. In this example, there are two runs to process. Use the spinner to set the count.
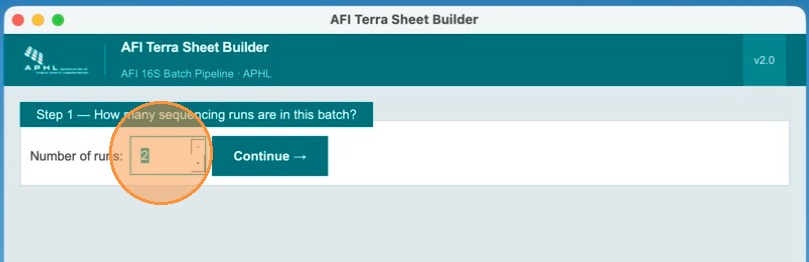
Click Continue
After setting the run count, click Continue to advance to the run-naming step.
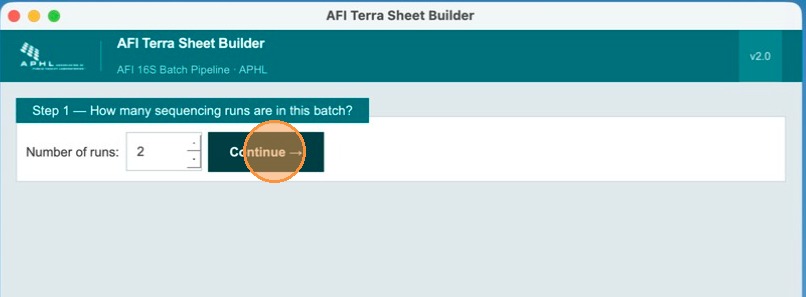
Name each run
In Step 2, enter a unique name for each run. Names are used as the run_id value for all samples in that run. No spaces are allowed — use underscores or hyphens (e.g., run_2024_11). In this example, runs are named Run 1 and Run 2. Click Next when done.
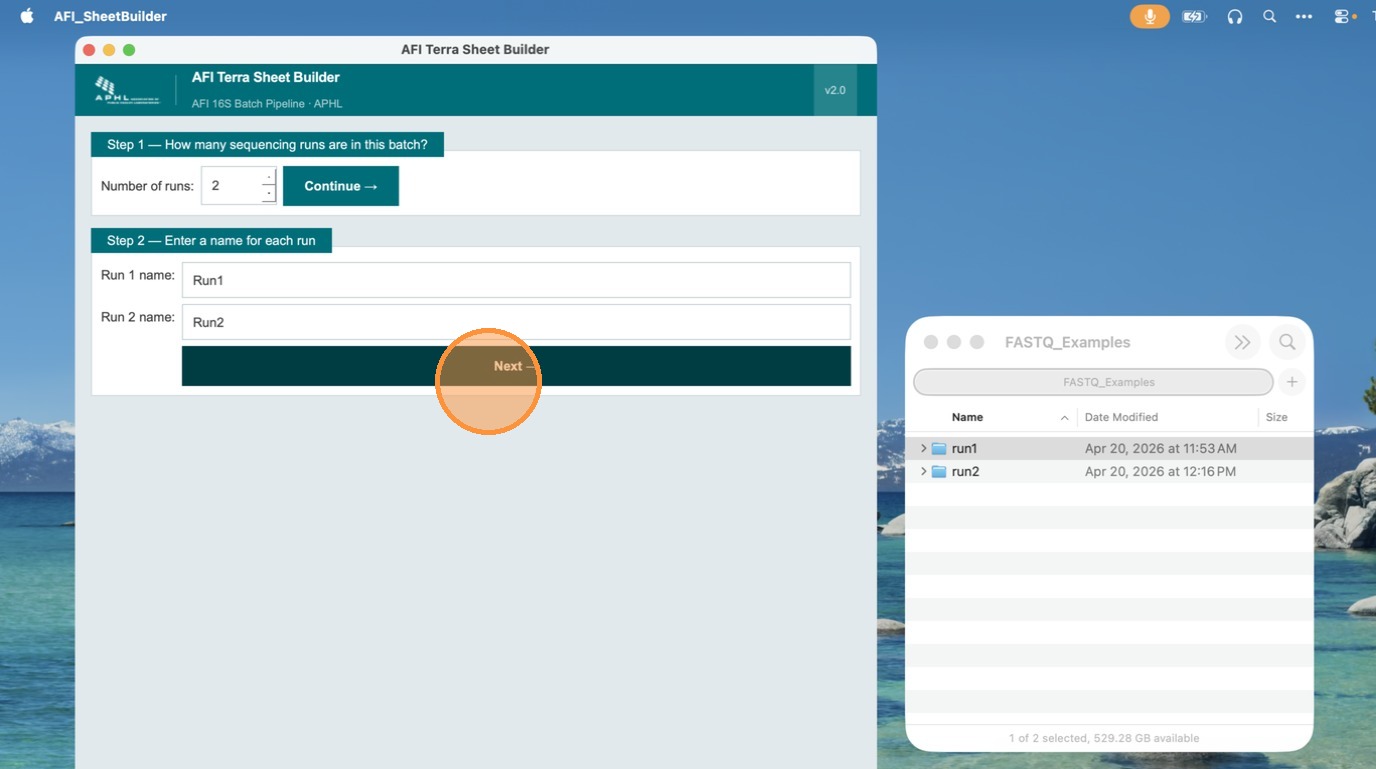
Select FASTQ files for each run
In this step, add FASTQ files to each run. You can import from pre-generated TSV files or browse to the folder where R1 and R2 FASTQ files are located. Click Browse Folder to select a directory — the app auto-discovers all R1/R2 pairs using the _R1_/_R2_ naming convention.
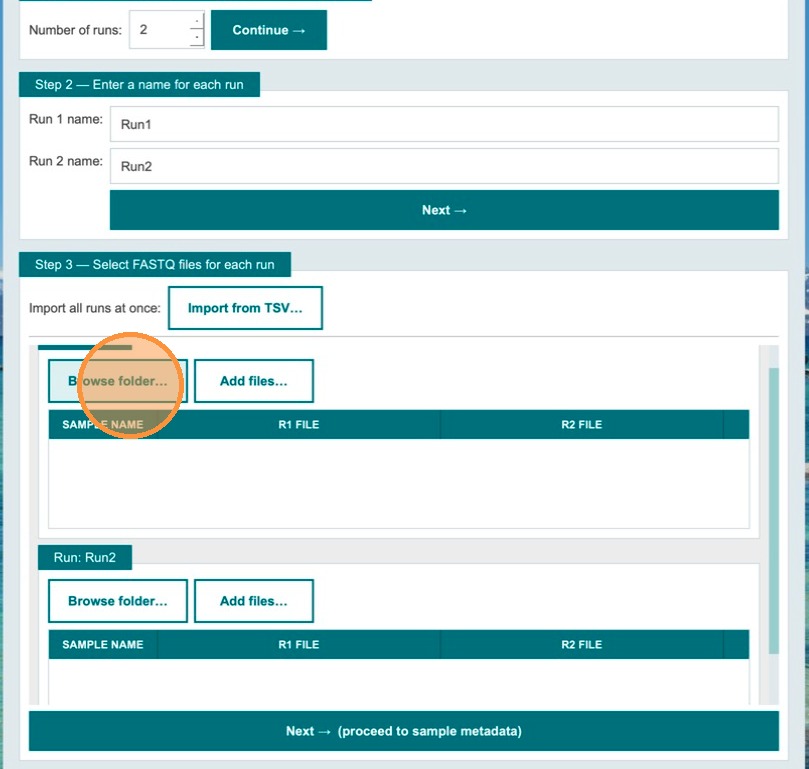
- Browse folder — auto-discovers all R1/R2 FASTQ pairs in a directory
- Add files… — multi-file dialog; app auto-pairs R1+R2 by name pattern
- Import from TSV… — global import with
run_idandsample_idcolumns
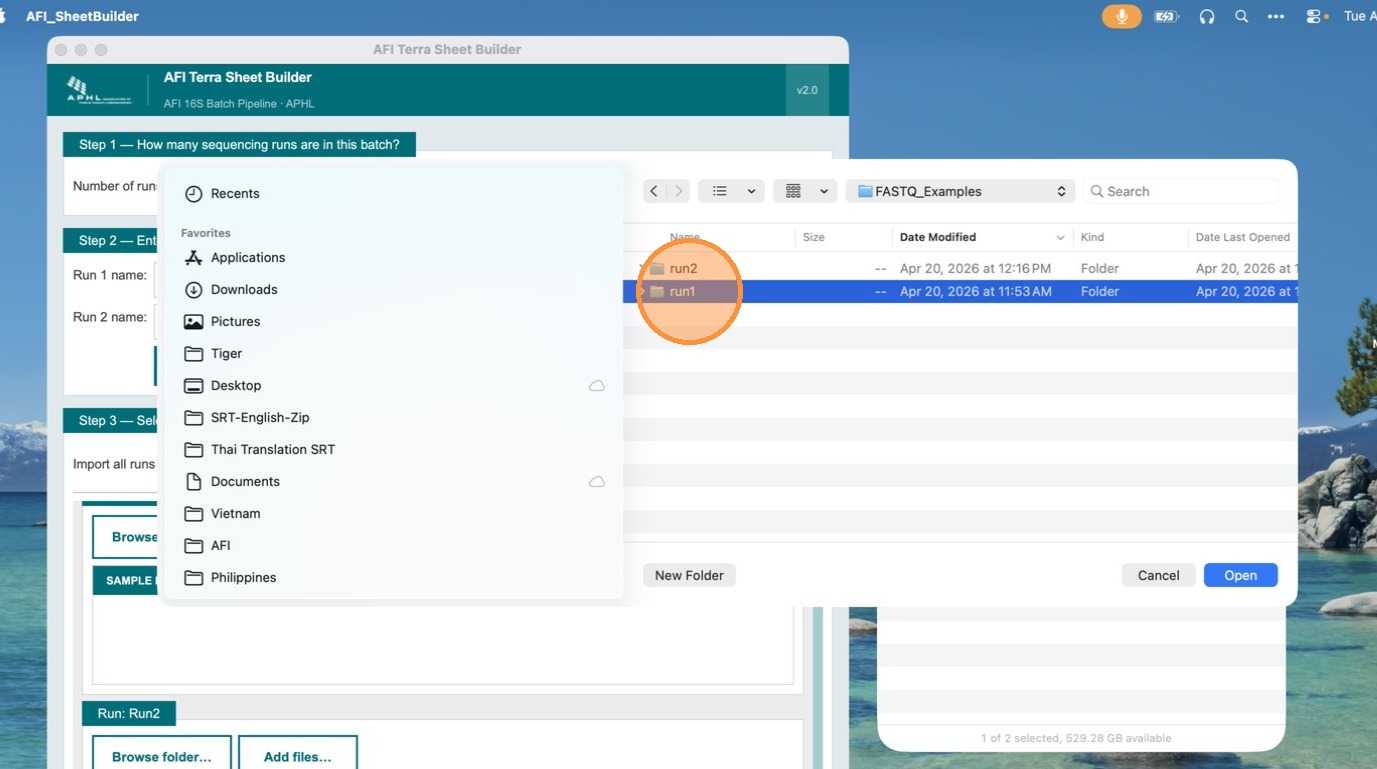
Browse to the FASTQ folder and click Open
Navigate to the folder containing the paired read files. Select the folder and click Open. The app will scan for all valid R1/R2 FASTQ pairs automatically.
Files are sorted into the metadata table automatically
The app sorts discovered files into the metadata table based on the specified runs. Repeat the folder selection for the remaining runs to be processed.
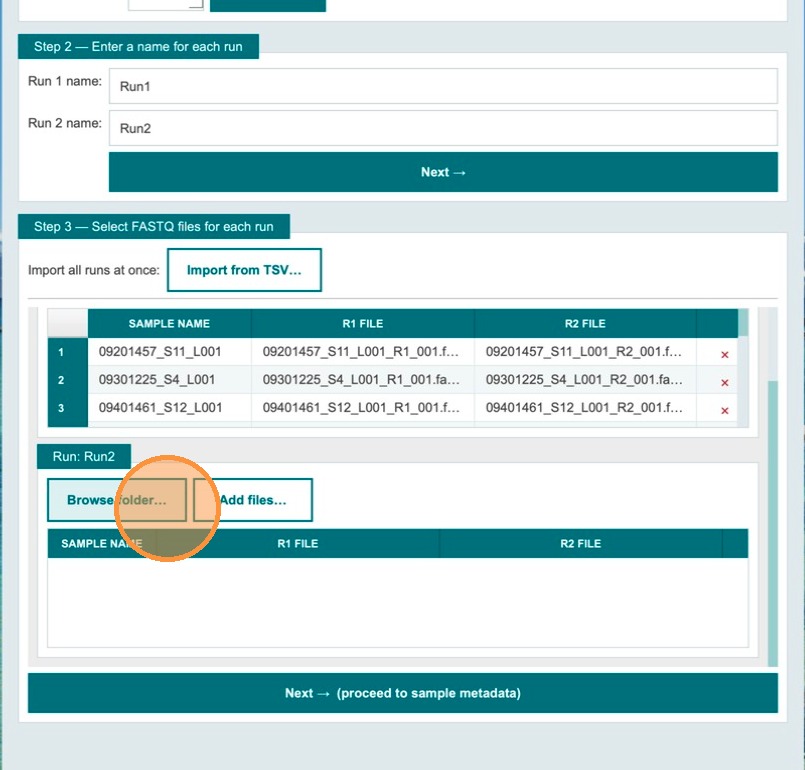
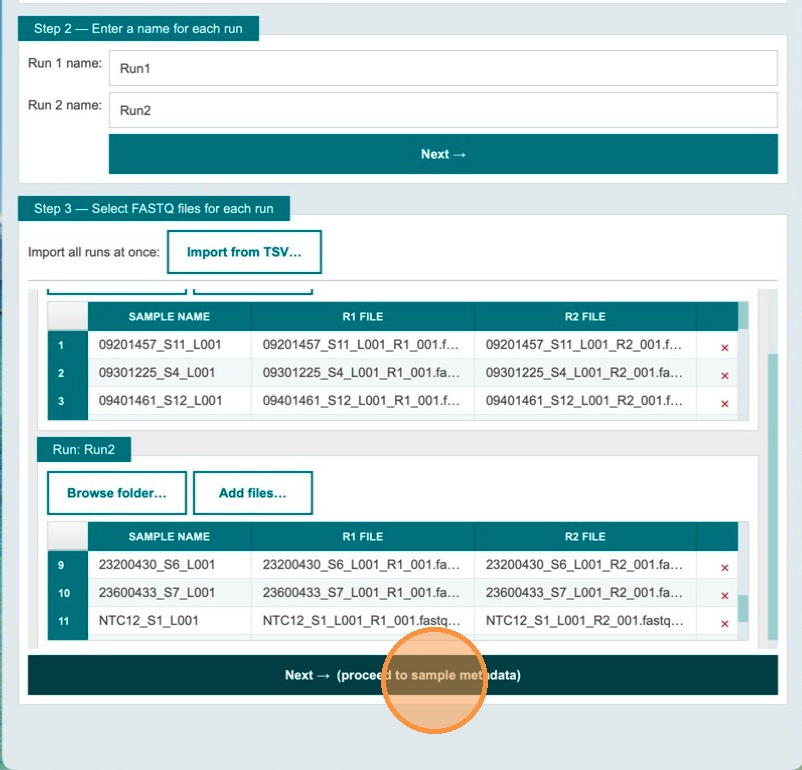
Verify sample names and file names, then click Next
Tables with sample names and file names are populated automatically. Review the table to confirm all entries are correct. Once ready, click Next to proceed to sample metadata entry.
Review the sample metadata page
This page requires additional information. Note that the Analysis Date field has already been pre-filled automatically with today's date.
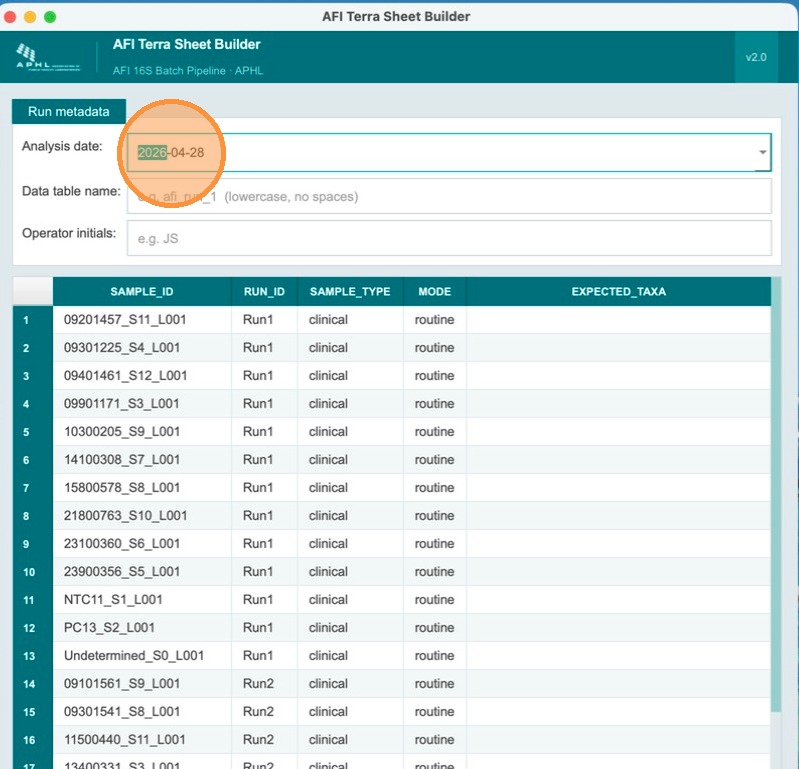
Enter the Terra table name and operator initials
Fill in the Terra metadata table name (lowercase alphanumeric, ≤32 chars, must start with a letter) and the operator's initials. These are recorded in the analysis_comments column of the output TSV for traceability.
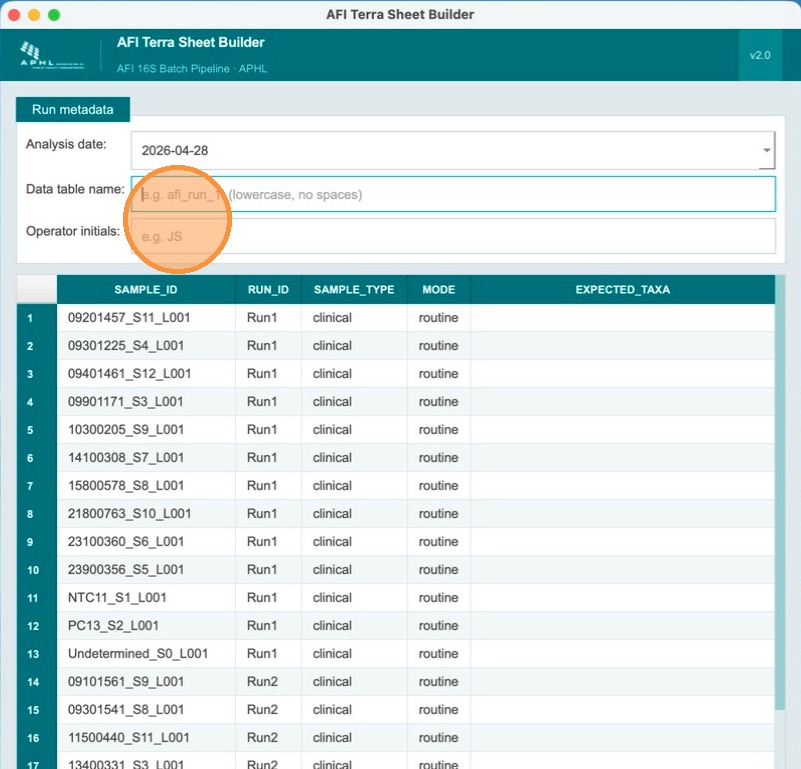
Designate the NTC (negative control) for each run
Below the header fields, the sample table lists all samples for both runs. Each run must have at least one NTC and one positive control. In the sample_type column, find the NTC sample for Run 1 and select NTC from the dropdown. The sample_type for clinical samples remains clinical.
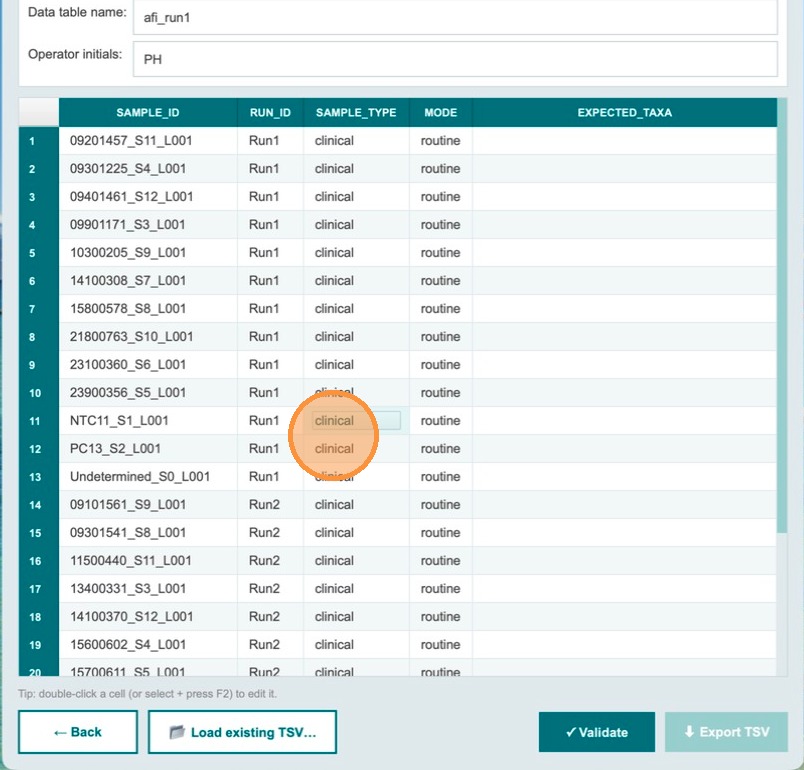
Confirm the NTC designation
The NTC sample for Run 1 is now designated. The sample_type dropdown shows NTC for that row. Continue to designate the positive control for Run 1.
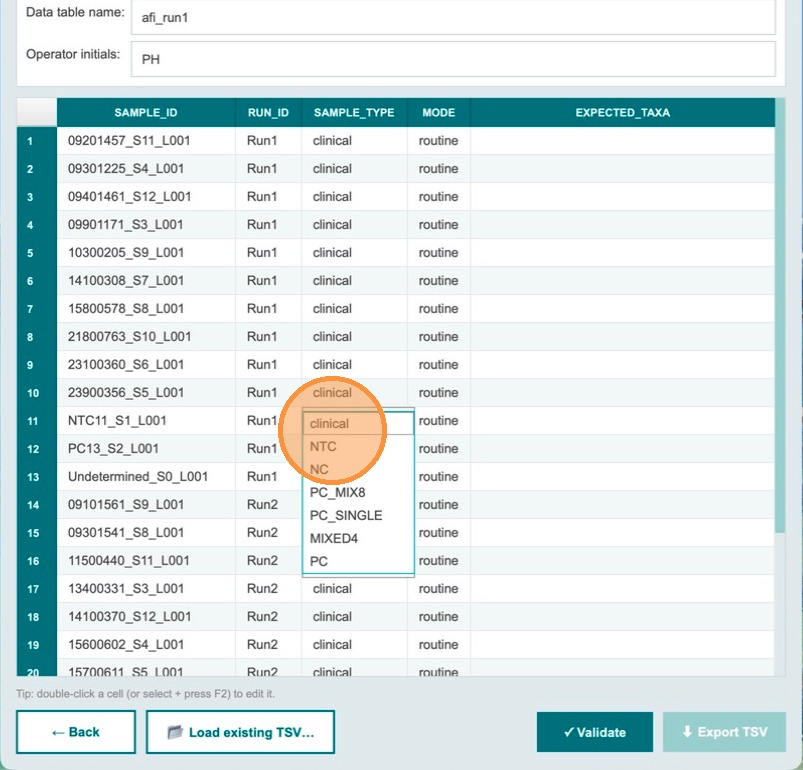
Begin changing the positive control sample type
For the positive control sample in Run 1, click the sample_type dropdown to change it from clinical to the appropriate control type.
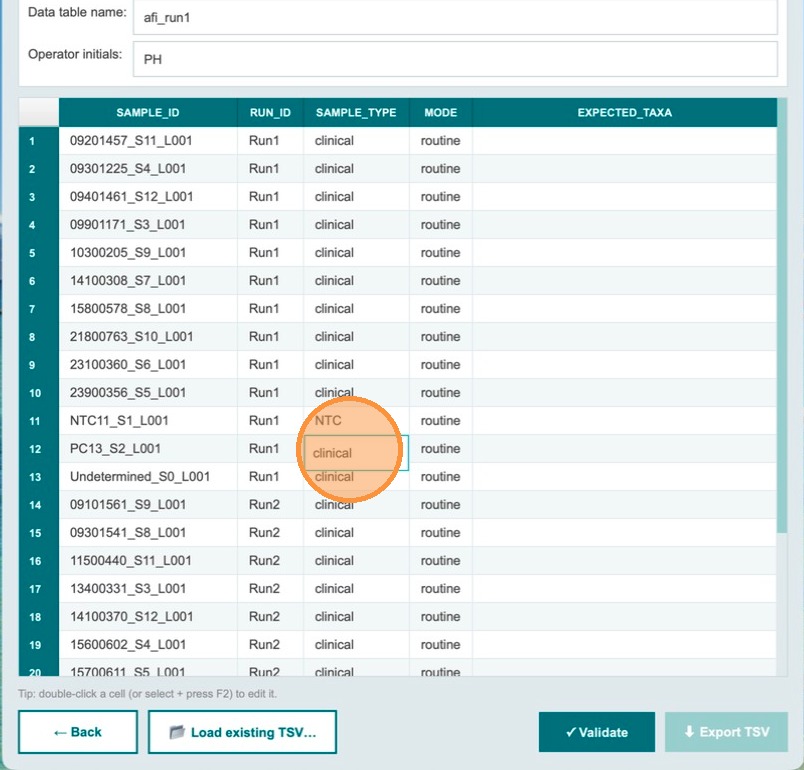
Open the sample type dropdown
Click the dropdown control for the positive control row to open the list of available sample types.
Select the positive control type
Change the sample_type to the type of control used. In this example, PC_MIX8 is selected — a positive control containing DNA from 8 bacterial species.
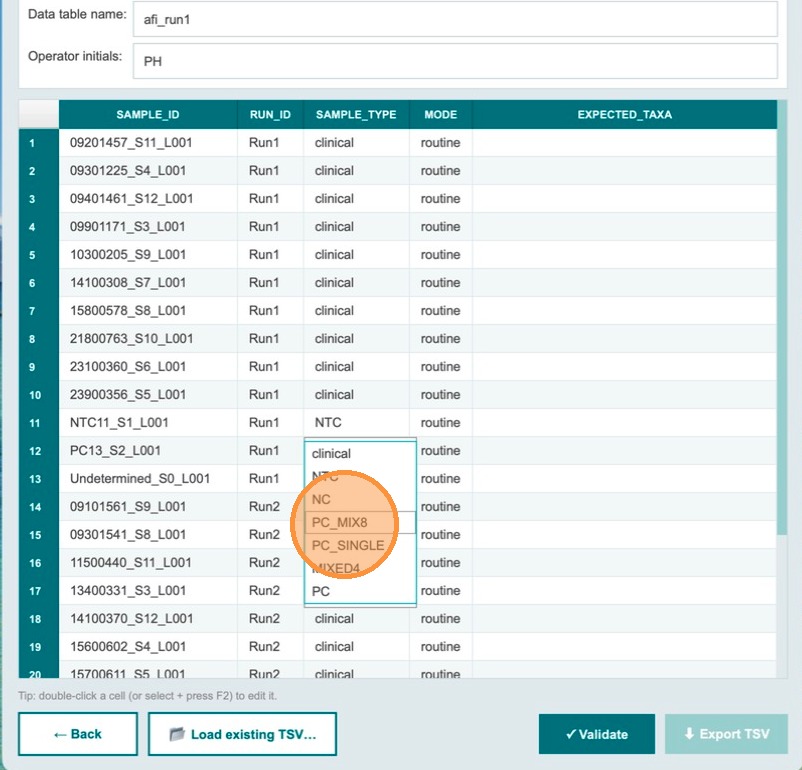
| sample_type | mode (auto-filled) | expected_taxa (auto-filled) |
|---|---|---|
PC_MIX8 | validation | Bacillus;Listeria;Staphylococcus;Enterococcus;Limosilactobacillus;Salmonella;Escherichia;Pseudomonas |
clinical | routine | (empty) |
NTC / NC | routine | (empty) |
Expected taxa auto-populate for PC_MIX8
Once PC_MIX8 is selected, the expected_taxa field auto-fills with the 8 bacterial species expected from that control. This tells the validation workflow which taxa should be identified.
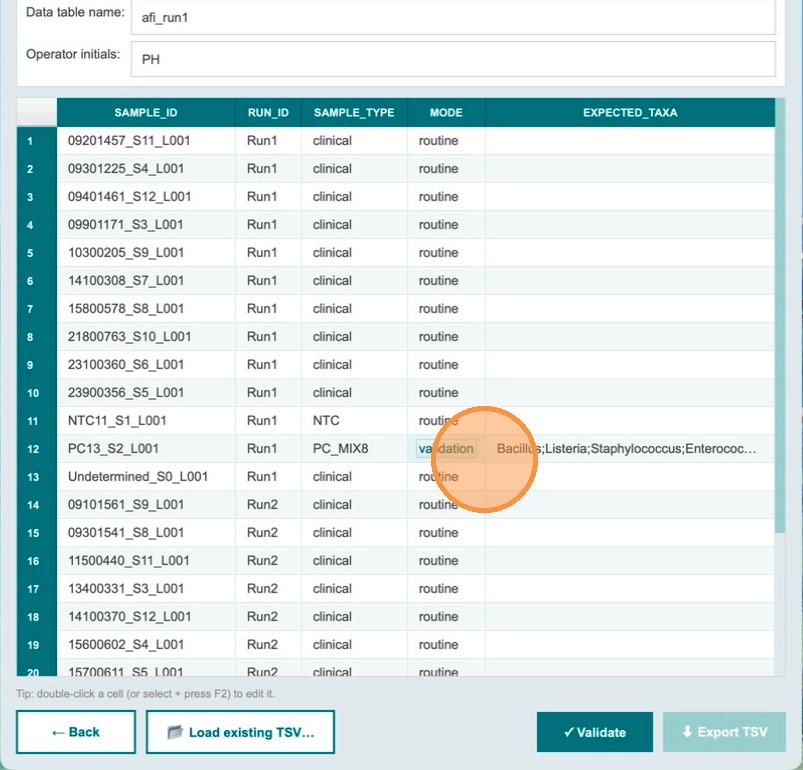
Validation catches missing controls before export
If an operator forgets to designate the NTC and positive control for Run 2 and clicks Validate, the Export TSV button remains greyed out. The app has detected missing required controls.
Review and fix the validation error
The app generates a descriptive error message stating that Run 2 has no NTC and no positive control specified. Click OK to acknowledge. Correct the missing designations for Run 2 before proceeding.
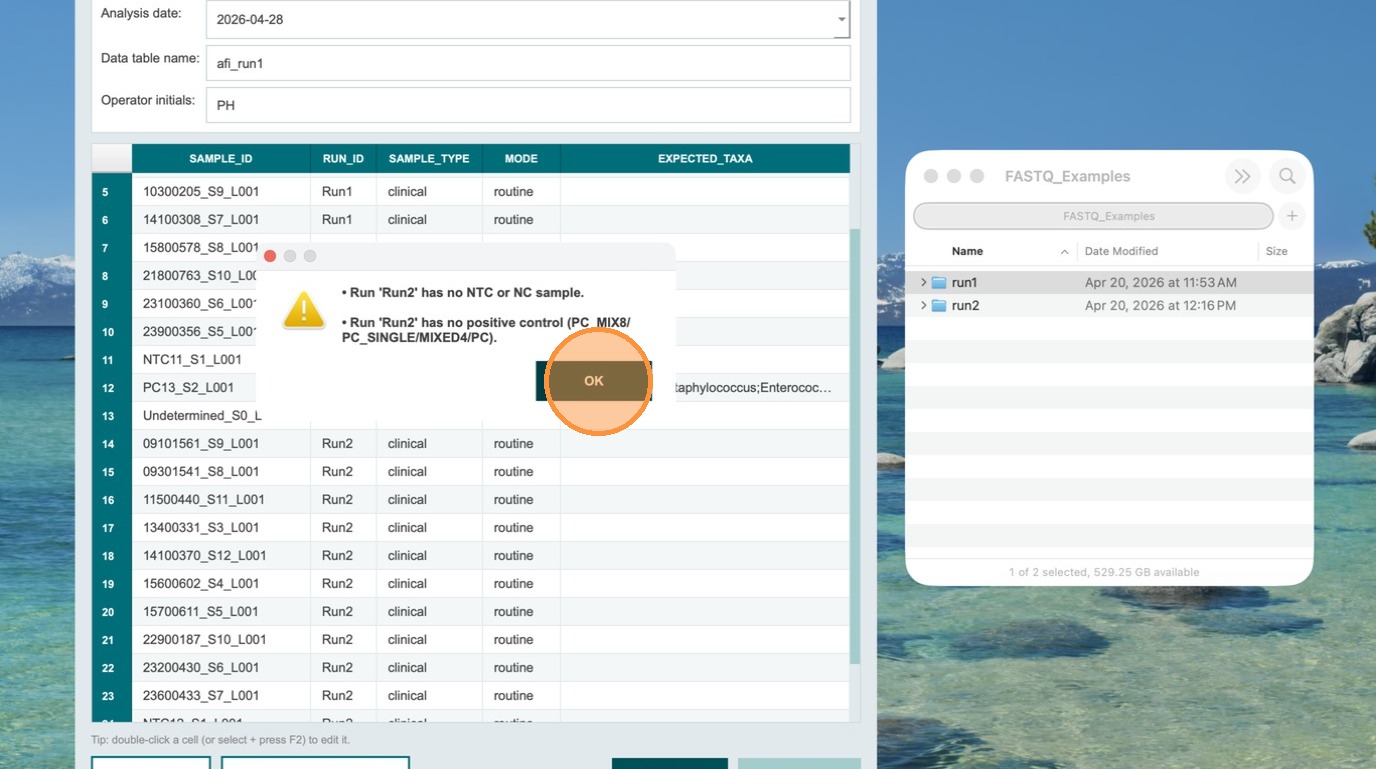
- No duplicate
sample_idvalues across all runs - Each run has at least one
NTCorNCsample - Each run has at least one positive control (
PC_MIX8,PC_SINGLE,MIXED4, orPC) - All validation-mode rows have a non-empty
expected_taxafield - Table name matches
^[a-z][a-z0-9_]{0,31}$
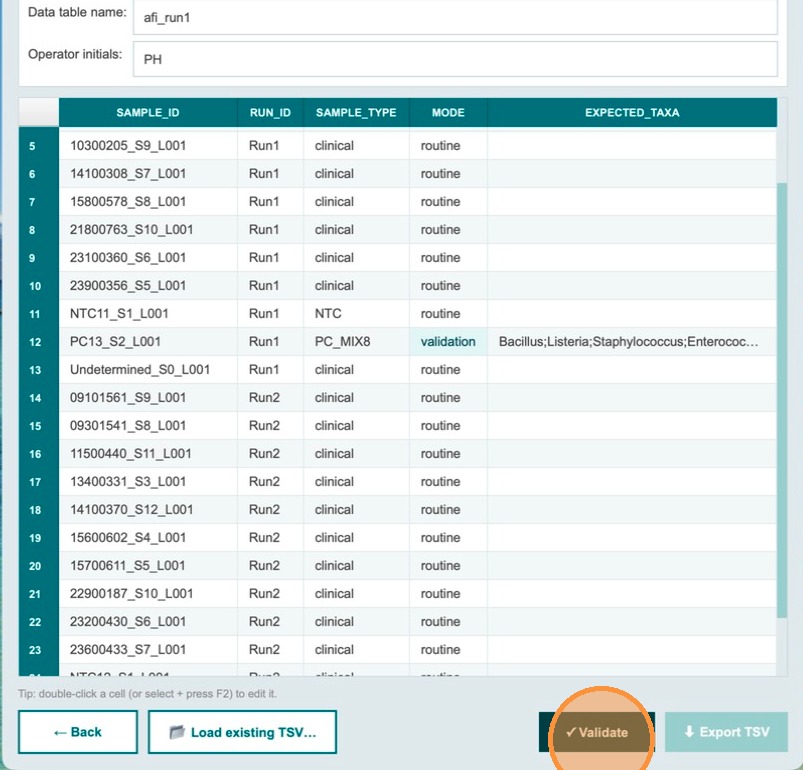
Validate — all checks must pass before export
After correcting all issues, click Validate again. If all checks pass, the Export TSV button becomes clickable. The TSV files are now ready to be uploaded to Terra.bio.
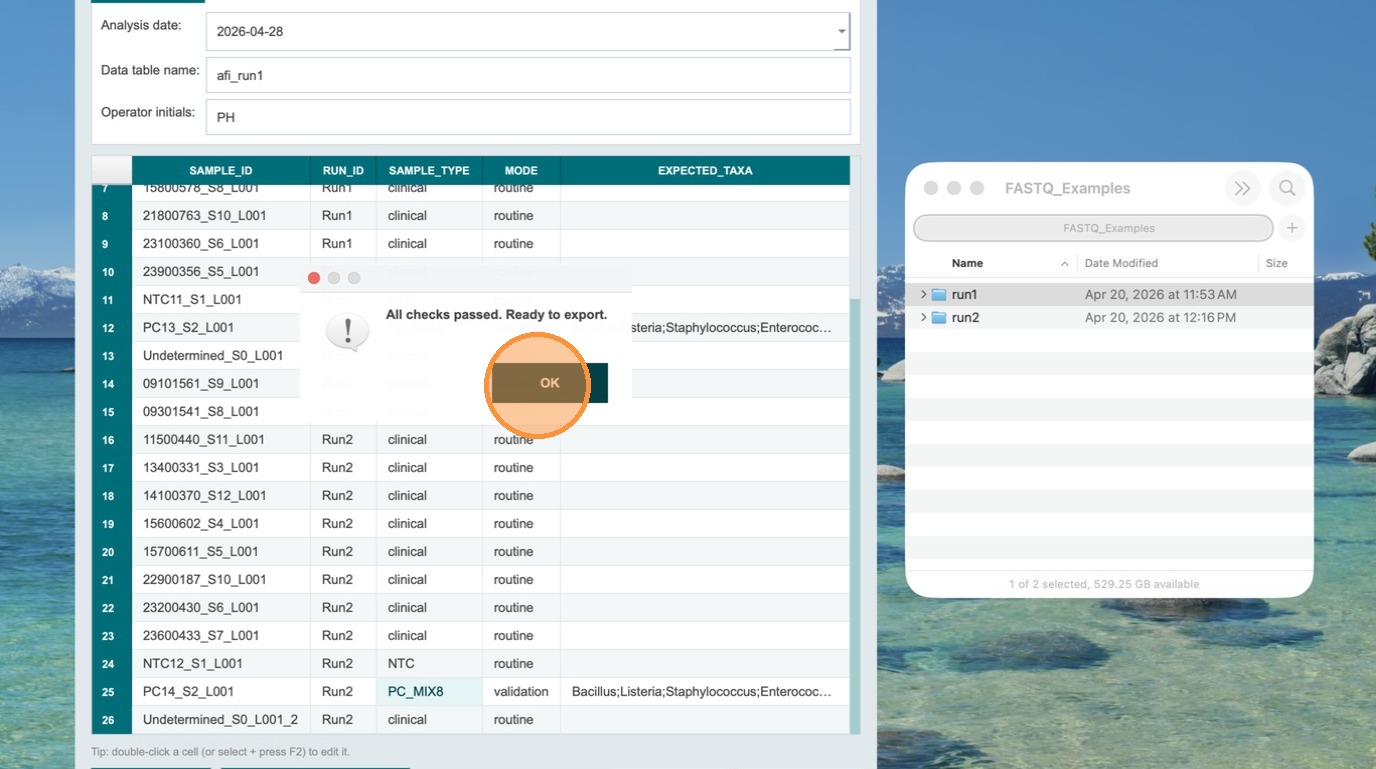
Export TSV and save the file
Click Export TSV and save the file to a location you can easily locate. The file will be named using the table name you entered in the header fields.
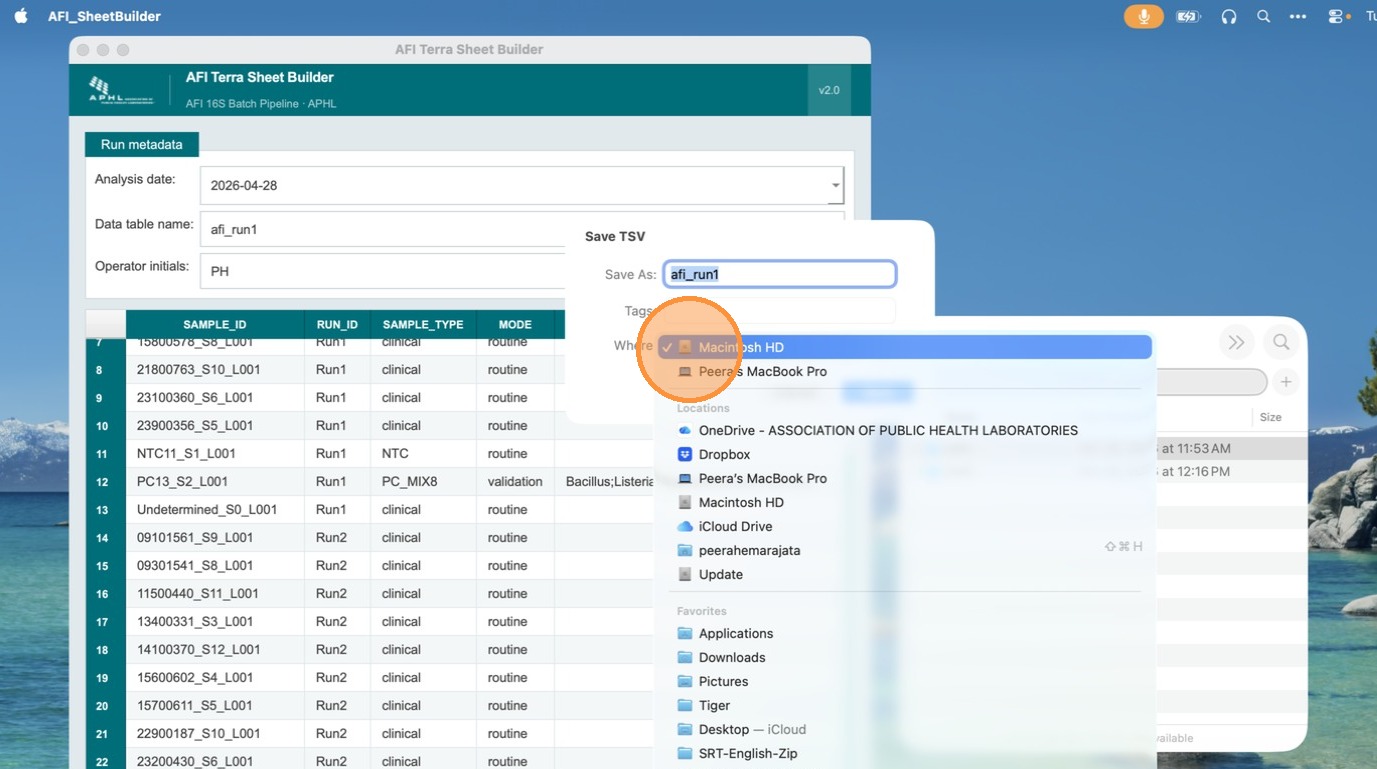
Verify the exported file in Microsoft Excel
Open the exported TSV in Microsoft Excel to confirm the format is correct and compatible with Terra.bio. Check that the entity:<table_name>_id column is the first column, all sample fields are present, and the analysis_comments column appears as the last column.
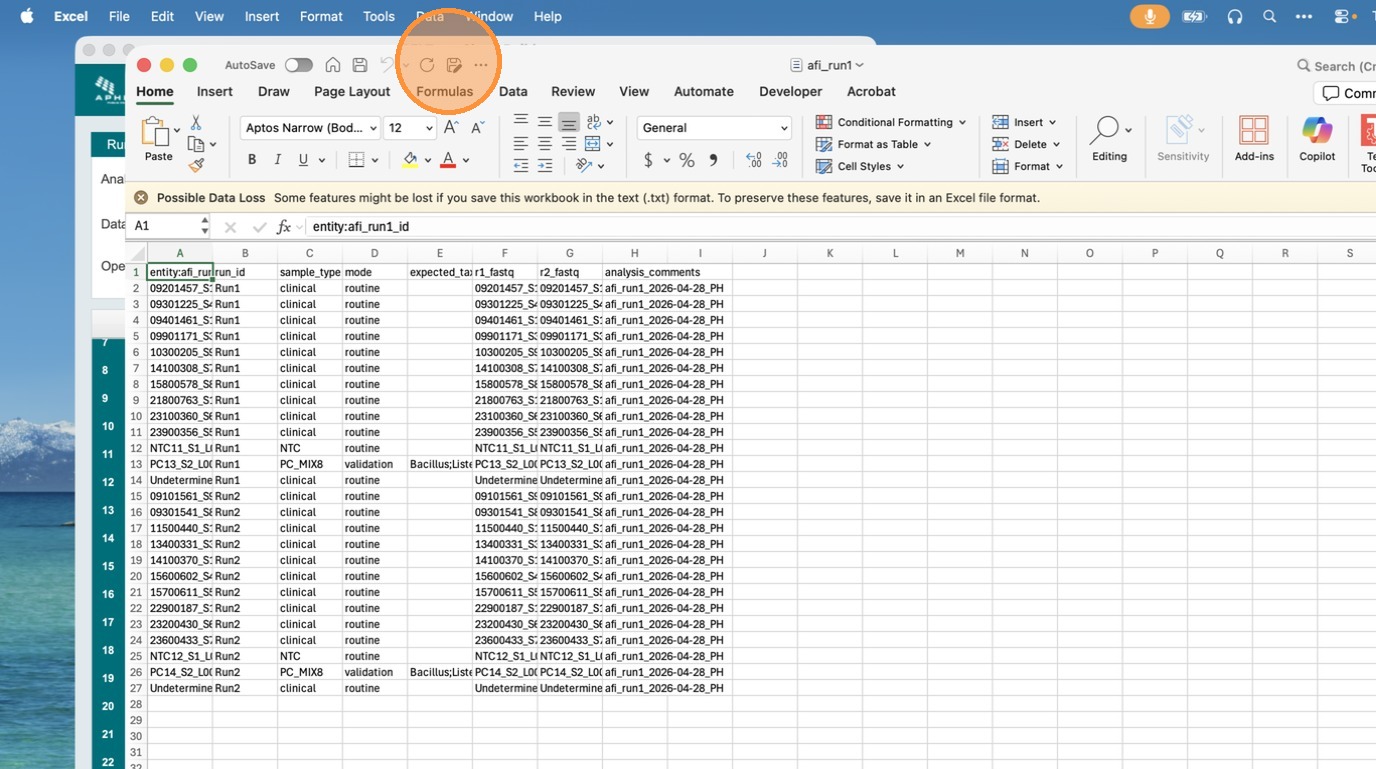
Importing the TSV into Terra
- In your Terra workspace, go to Data → Import Data → Upload TSV.
- Select the exported TSV file.
- Terra will create or update a sample set entity table with the name you provided.
The exported file uses entity:<table_name>_id as the first column (required by Terra) followed by all sample fields. An analysis_comments column appended as the last column contains <table_name> | <date> | <initials> for traceability.
9. Helper Scripts
All helper scripts are in the scripts/ directory. They run locally (not in Terra) and are used to prepare inputs, post-process outputs, and manage Docker images.
build_batch_inputs_json.py
Converts a sample sheet TSV or AFI mapping TSV into a Terra-ready batch input JSON.
python3 scripts/build_batch_inputs_json.py \ --sample-sheet wdl/inputs/batch_samples.template.tsv \ --out-json wdl/inputs/batch_run1.json \ --rickettsiales-panel gs://YOUR_BUCKET/ref/rickettsiales_panel_16S.clean.fa \ --centrifuger-db centrifuger_bact_arch_plus_rickettsiales
build_ntc_background_from_metrics.py
Builds a run-specific NTC background TSV from one or more NTC align_metrics.tsv output files.
python3 scripts/build_ntc_background_from_metrics.py \ --ntc-metrics path/to/NTC1.align_metrics.tsv \ --ntc-metrics path/to/NTC2.align_metrics.tsv \ --out wdl/inputs/run_ntc_background.tsv
make_terra_import_sheet.py
CLI companion to the Terra Sheet Builder GUI. Generates or validates a Terra-compatible sample import TSV and optionally writes an annotated Excel workbook.
python3 scripts/make_terra_import_sheet.py --template --output my_run.tsv python3 scripts/make_terra_import_sheet.py --input samples.csv --output terra_import.tsv --excel
10. Output Files Reference
Per-sample outputs
| File | Description |
|---|---|
<id>.genus_counts.tsv | Genus-level Centrifuger read counts |
<id>.bam / .bai | minimap2 alignment, sorted and indexed |
<id>.align_metrics.tsv | Per-genus alignment metrics (mapped_reads, max_breadth) |
calls.tsv | NTC-aware taxa calls — the core interpretive output |
taxa_evidence.tsv | Detailed per-genus evidence with justification strings |
routine_summary.tsv | Detected taxa list (routine mode only) |
validation_summary.tsv | Concordance summary (validation mode only) |
Batch-only outputs
| File | Description |
|---|---|
ntc_background_<run_id>.tsv | Auto-computed NTC background per distinct run_id |
run_summary.tsv | Batch-level summary with per-run PC8 validity — one row per sample |
11. Interpreting Calls and Summaries
Call values — alignment source (Orientia and Rickettsia)
| Call | Meaning |
|---|---|
| Confirmed | Strong positive: reads and breadth meet confirmation thresholds and are well above NTC |
| Probable | Moderate positive: lower thresholds met and reads exceed NTC |
| Not_Confirmed | Equivocal: sufficient reads but at or below NTC level; cannot distinguish from noise |
| Negative | Insufficient reads |
Call values — Centrifuger source (all other genera)
| Call | Meaning |
|---|---|
| Detected | Reads exceed floor threshold and are well above NTC |
| Not_Detected | Negative or below threshold |
PC8 validity
For PC_MIX8 samples, a pass/fail flag is computed. At least 6 of the 8 expected taxa must be detected for a passing result. In run_summary.tsv, run_pc8_valid carries this value for every row, allowing immediate run-level QC assessment.
12. Detection Thresholds
Configurable thresholds (Tier 1 "Confirmed" and Centrifuger "Detected")
| Input | Default | Description |
|---|---|---|
align_confirm_reads | 100 | Minimum mapped reads for Confirmed alignment call |
align_confirm_breadth | 0.25 | Minimum breadth of coverage for Confirmed call |
align_fold | 5.0 | Minimum fold over NTC reads for Confirmed call |
cfr_floor | 500 | Minimum Centrifuger reads for Detected call |
cfr_fold | 5.0 | Minimum fold over NTC reads for Detected call |
Hardcoded thresholds (Tier 2 "Probable" and equivocal calls)
The Tier 2 order-level Rickettsiales rescue and the equivocal-call thresholds are defined in scripts/call_taxa.py. These are not currently exposed as workflow inputs but are documented here for transparency.
| Call | Reads | Breadth | NTC requirement |
|---|---|---|---|
| Confirmed | ≥ align_confirm_reads (100) | ≥ align_confirm_breadth (0.25) | ≥ align_fold × NTC reads (5×) |
| Probable | ≥ 50 | ≥ 0.20 | > NTC reads (no fold requirement) |
| Not_Confirmed | ≥ 50 | (any) | ≤ NTC reads |
| Negative | < 50 | — | — |
13. V4 Decontamination Filter (post-pipeline)
.calls.tsv outputs and does not modify pipeline behavior or thresholds.
The filter is implemented as a standalone Python script (afi_decontamination_filter_v4.py) that ingests .calls.tsv rows with positive calls (Detected / Confirmed / Probable), applies the tiered rules below, and emits a report of which detections were kept and which were removed.
Tier A — High-confidence kit / skin / water contaminants
The following 11 genera are removed on detection in clinical and study samples. Membership is documented in five or more independent low-biomass contamination reviews and confirmed in this dataset's NTC profiles.
| Genus | Most common source |
|---|---|
| Pseudomonas | Kits, water, Taq, airborne |
| Ralstonia | Kits, ultrapure water biofilm |
| Bradyrhizobium | Kits, ultrapure water |
| Sphingomonas | Kits, water systems |
| Stenotrophomonas | Kits, water, PCR reagents |
| Methylobacterium | Kits, ultrapure water systems |
| Acinetobacter | Kits, PCR reagents, foot traffic |
| Cutibacterium | Skin (alcohol-resistant), airborne |
| Staphylococcus | Skin, airborne (coagulase-negative spp.) |
| Corynebacterium | Kits, skin, airborne |
| Brevundimonas | Kits, water; up to 285,740 NTC reads observed in run 6_and_7 |
Tier A exception — Burkholderia species-level safeguard
The genus Burkholderia is a Tier-A-equivalent kit contaminant (the genus is dominated in low-biomass samples by B. cepacia complex species), but it also contains B. pseudomallei, the etiologic agent of melioidosis — a "cannot-miss" diagnosis endemic in Thailand. For every sample with a Burkholderia genus detection, the filter:
- Opens the Centrifuger kreport (
<sample>.centrifuger.kreport.tsv) and parses rows at the species rank (S). - Sums reads assigned to Burkholderia pseudomallei (NCBI taxonomy ID 28450) at species rank.
- Retrieves the run's NTC species-level B. pseudomallei read count for comparison.
- Preserves the detection only if both: (i) species reads ≥ 500, AND (ii) species reads > the run-NTC species max.
- When preserved, the retained record stores the species-level read count, not the genus total.
pseudomallei_group and then erroneously reported the genus-level Burkholderia read count. In one study sample, this conflated 54,126 genus-level reads (dominated by B. cepacia complex species) with only 8 reads of B. pseudomallei at species rank. V4 parses the species rank correctly and gates preservation on species reads ≥ 500 against the same-run NTC species count.
Tier B — NTC-only organisms
Nine genera observed only in NTC samples across all sequencing runs are removed globally: Cereibacter, Thioclava, Bdellovibrio, Saltatorellus, Pseudogemmobacter, Minisyncoccus, Rhodoluna, Microbacterium, Arcanobacterium.
Tier 1 — Ultra-low-abundance environmental noise
Sixteen genera with median per-sample abundance < 0.5% across the dataset are removed: Shigella, Metapseudomonas, Stutzerimonas, Capsulimonas, Chamaesiphon, Chloroflexus, Flavihumibacter, Hymenobacter, Limnoglobus, Methylovirgula, Microvirga, Pelagovum, Pseudonocardia, Rufibacter, Salmonella, Spirosoma.
Tier 2 — Marginal organisms (conditional removal)
Twenty-seven genera with median 0.5–2.0% abundance are retained only if detected in ≥ 2 samples AND each detection is ≥ 1.0% abundance. Below those thresholds, they are removed as likely contaminants.
Positive-control spike-in bypass
For samples designated as positive controls, Tier A removal is bypassed for any organism that is a documented spike-in for that sample. Without this bypass, the P-aeru_S5_L001 PC would fail because Pseudomonas is in Tier A — but Pseudomonas aeruginosa is precisely the organism the PC is designed to detect. The bypass restores PC interpretability without weakening Tier A removal in clinical samples.
| PC sample | Type | Expected genera (filter bypassed for these) |
|---|---|---|
E-coli_S4_L001 | PC_SINGLE | Escherichia |
P-aeru_S5_L001 | PC_SINGLE | Pseudomonas |
S-pneumo_S2_L001 | PC_SINGLE | Streptococcus |
S-suis_S3_L001 | PC_SINGLE | Streptococcus |
PC_MIX8 replicates (5) | PC_MIX8 | Bacillus, Enterococcus, Escherichia, Limosilactobacillus, Listeria, Pseudomonas, Salmonella, Staphylococcus (ZymoBIOMICS Microbial Community Standard) |
Mixed_S6_L001 | MIXED4 | Escherichia, Pseudomonas, Streptococcus |
Under the bypass, PC concordance is defined as all expected spike-in organisms must be detected AND retained by the filter.
Running the filter
python3 afi_decontamination_filter_v4.py
The script reads .calls.tsv and Centrifuger kreport files from a configured run directory and writes DECONTAMINATION-FILTER-REPORT-V4.txt with a per-sample / per-detection breakdown of filter actions. A companion appendix generator (generate_appendices.py) produces Markdown and Excel tables with biomass distribution, NTC cross-checks, and confidence labels.
14. Analytical Validation Summary
Validation panel composition
| Category | n | Description |
|---|---|---|
| Clinical (reference-lab confirmed) | 33 | E. coli (5), O. tsutsugamushi (6), Rickettsia (4), Leptospira (4), B. pseudomallei (5), S. pneumoniae (3), S. suis (3), C. burnetii (2), Yersinia (1) |
| Positive controls (PC_SINGLE / PC_MIX8 / MIXED4) | 10 | 4 PC_SINGLE, 5 PC_MIX8 (ZymoBIOMICS Standard), 1 MIXED4 |
| Negative template controls (NTC) | 5 | Water through extraction + library prep |
| Total | 48 | Across 9 sequencing runs |
Sample-level analytical performance
| Category | Concordant | Total | Rate | 95% CI |
|---|---|---|---|---|
| Clinical (target detected AND retained by V4) | 21 | 33 | 63.6% | 46.6–77.8% |
| Positive controls (all expected spike-ins detected + retained) | 10 | 10 | 100% | 72.2–100% |
| NTCs (no TAC bacterial target genus retained) | 5 | 5 | 100% | 56.6–100% |
| Sample-level analytical performance (clinical + PC) | 31 | 43 | 72.1% | 57.3–83.3% |
| Overall validation accuracy | 36 | 48 | 75.0% | 61.2–85.1% |
The 72.1% sample-level analytical performance is the appropriate headline figure for regulatory documentation and matches the legacy APHL bioinformatic validation report for the same panel.
AFI study cohort (n=86)
- 71 / 86 samples (82.6%) carried ≥ 1 positive call (Centrifuger Detected, Tier 1 Confirmed, or Tier 2 Probable).
- 15 / 86 samples (17.4%) had zero detected taxa — consistent with pre-sequencing failure (DNA extraction, library prep, or sequencing depth).
- V4 removed 70 / 217 detections (32.3%): predominantly Cutibacterium, Staphylococcus, Brevundimonas, Acinetobacter, Corynebacterium.
- 147 detections retained across the cohort.
Key cohort findings
16901195_S5_L001, breadth 0.3235, ~12× NTC headroom — the highest-confidence Rickettsiales call in the cohort), and ten Tier 2 order-level "Rickettsiales detected" rescues (breadth 0.21–0.24). This pattern aligns with the expected endemic epidemiology of northeastern Thailand, where Rickettsiales (obligate intracellular, cannot be cultured on routine blood agar) account for a substantial fraction of AFI presentations. All 11 samples warrant species-specific qPCR confirmation.
Additional candidate detections (require orthogonal confirmation): Leptospira in one sample at 22.78% (with a same-run NTC contamination caveat — the NTC carried 78,691 Leptospira reads, ~10× the study sample); Brucella in three samples at near-noise abundance (mean 0.98%, max 1.96%); Streptococcus in five samples (V1–V3 cannot resolve species).
Companion documents (manuscript-ready package)
| File | Content |
|---|---|
MANUSCRIPT-FINAL-DRAFT.md | Long-form manuscript draft (~7,800 words): Title, Abstract, Introduction, Methods, Results, Discussion, Limitations, Conclusion, References, Appendices |
MANUSCRIPT-CONDENSED-DRAFT.md | JCM-style condensed draft (~3,900 words) |
MANUSCRIPT-WALKTHROUGH-THAI.md | Thai-language walkthrough for the AFI wet-lab team |
APPENDIX-VALIDATION-PANEL.md / APPENDIX-STUDY-SAMPLES.md | Per-sample detection tables (Markdown) |
APPENDICES.xlsx | Same per-sample tables in Excel (4 sheets: validation detections, validation summary, study detections, study summary) |
figure_a_sankey.html / figure_a_sankey.png | Figure 1: Sankey diagram of pre- vs. post-V4 filter genus distribution |
DECONTAMINATION-FILTER-REPORT-V4.txt | Raw V4 filter output with summary statistics and Burkholderia species evidence per sample |
Methodological revision log
The following corrections were identified during a critical review and are reflected throughout this documentation and the manuscript drafts:
| Item | Pre-revision (V3) | Post-revision (V4) |
|---|---|---|
| B. pseudomallei safeguard | Substring match on "pseudomallei" → reported genus-level read count | Species-rank kreport parsing with ≥ 500 read floor and NTC comparison |
| Mycoplasmopsis | Removed under Tier 1 "ultra-low abundance" (mis-categorized) | Retained (mean 39.78% across 4 samples) |
| Brevundimonas | Not in any tier; retained at mean 16% across 9 samples | Added to Tier A; removed |
| Study cohort size | "56 samples" (counted only Centrifuger-Detected entries) | 86 samples (71 with detections, 15 zero-detection pre-seq failures) |
| Rickettsiales in cohort | "0 samples" (Centrifuger-only view) | 11 samples (with Minimap2 rescue rows included) |
| Sample 00618_S7_L001 | "Failed rescue → Discordant" | Tier 2 Probable rescue → Concordant |
| P-aeru_S5_L001 PC | Failed (Pseudomonas removed by Tier A) | Passes with PC spike-in bypass |
| Reference citations (Glassing, Lauder, Tan) | Garbled author names | Verified against PubMed (PMID 27239228, 27338728, 36997797) |
15. Docker Images
| Input | Default image | Description |
|---|---|---|
afi_core_docker | phemarajata614/afi-terra:0.4.1 | Core analysis image (Python, samtools, minimap2) |
fastp_docker | staphb/fastp:0.23.4 | QC trimming |
centrifuger_docker | phemarajata614/centrifuger:1.1.0 | Centrifuger classifier |
Building and pushing the AFI core image
bash scripts/build_push_afi_core_image.sh phemarajata614 0.4.1 linux/amd64
16. Troubleshooting
The default Terra VM is too small. Set
centrifuger_memory = "96G" and centrifuger_disks = "local-disk 375 HDD" in your input JSON.
Every
run_id in the batch must include at least one sample with sample_type of NTC or NC. Check the Sheet Builder validation output for missing controls.
Do not upload only the main WDL file. Either use Dockstore (recommended) or package all WDL files and task imports into a ZIP archive before uploading.
Click Validate first. The button enables only after all validation checks pass. Review the error messages and correct missing controls, duplicate sample IDs, or invalid table name format.